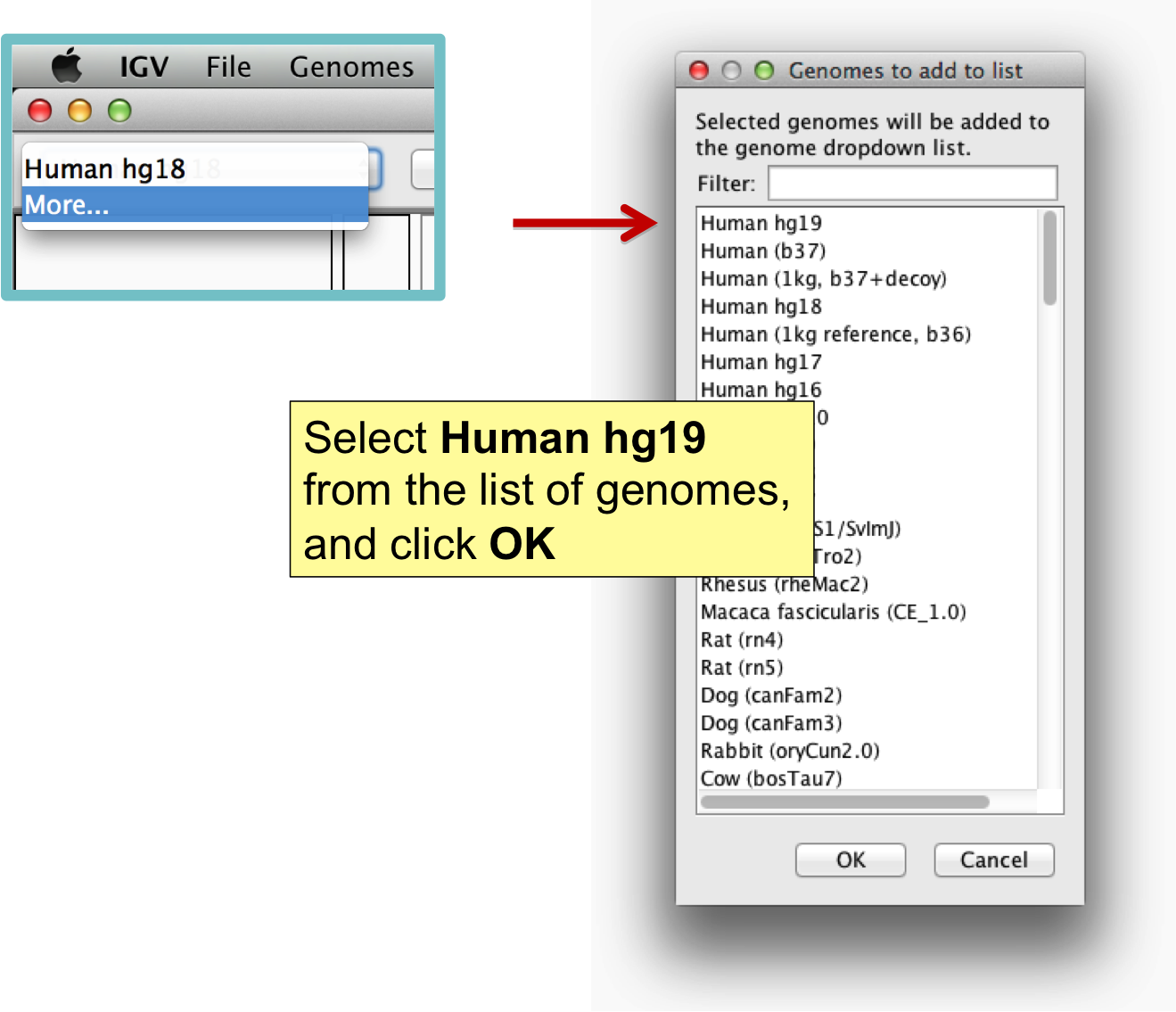
1. Installing Integrative Genomics Viewer
Launch IGV from website: IGV Download Site
Latest Version: 2.3.4
Launch IGV from website: IGV Download Site
Latest Version: 2.3.4
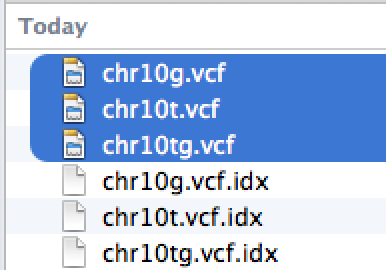
| File | Description | Type |
|---|---|---|
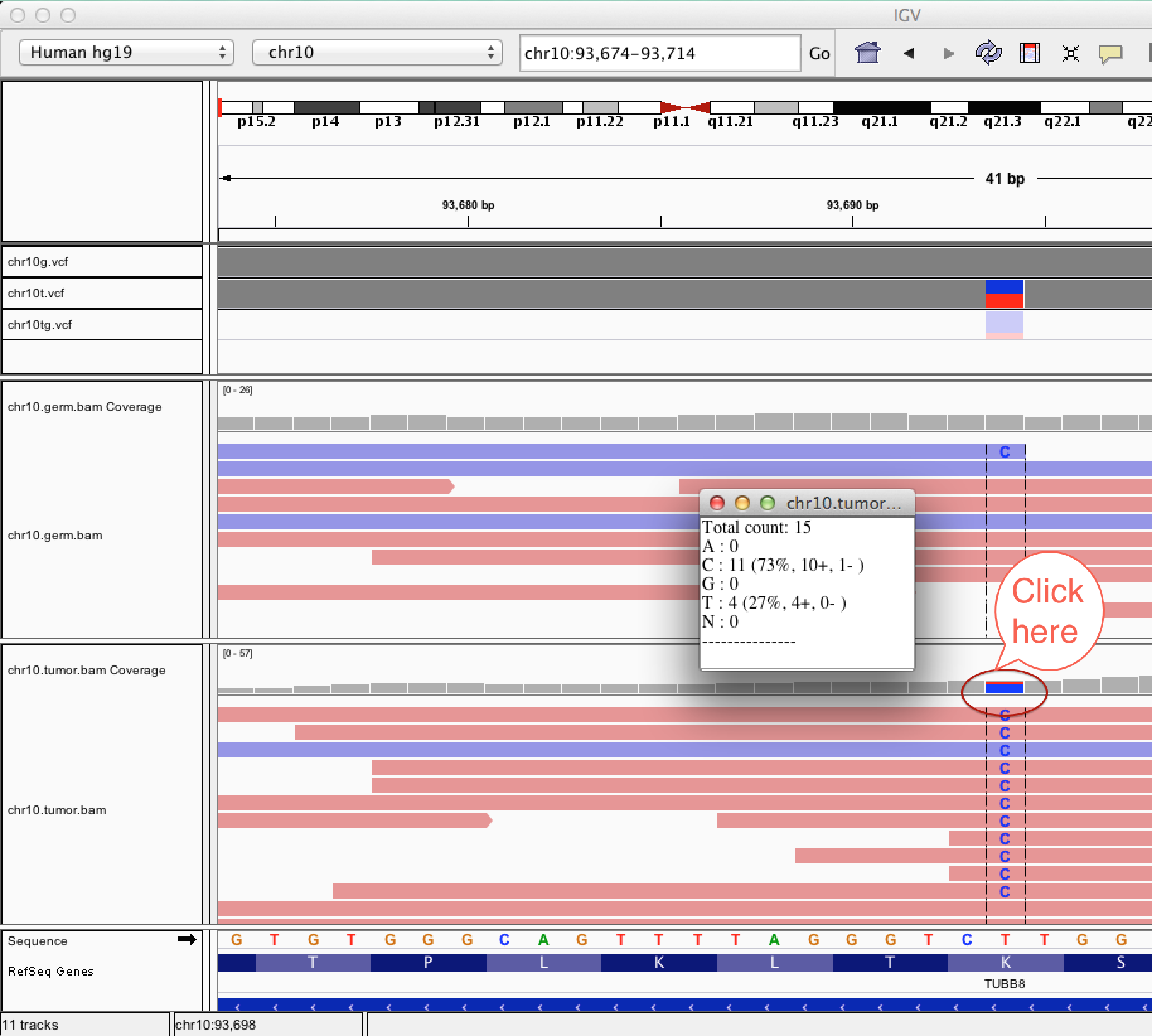
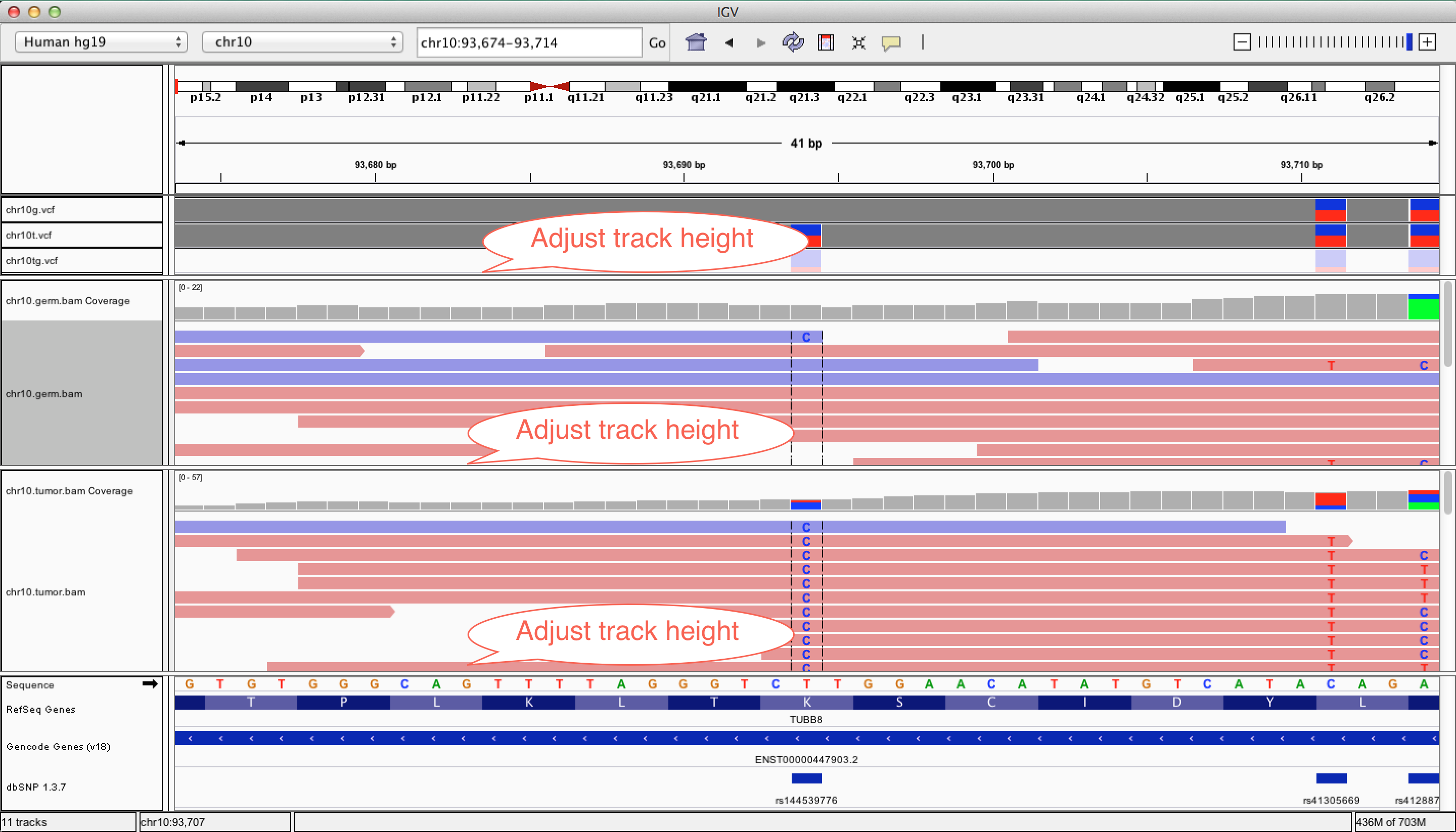
| chr10g.vcf | Normal (germline) sample vcf produced by germline variant caller (HaplotypeCaller, GATK) | vcf |
| chr10t.vcf | Tumor sample vcf produced by germline variant caller (HaplotypeCaller, GATK) | vcf |
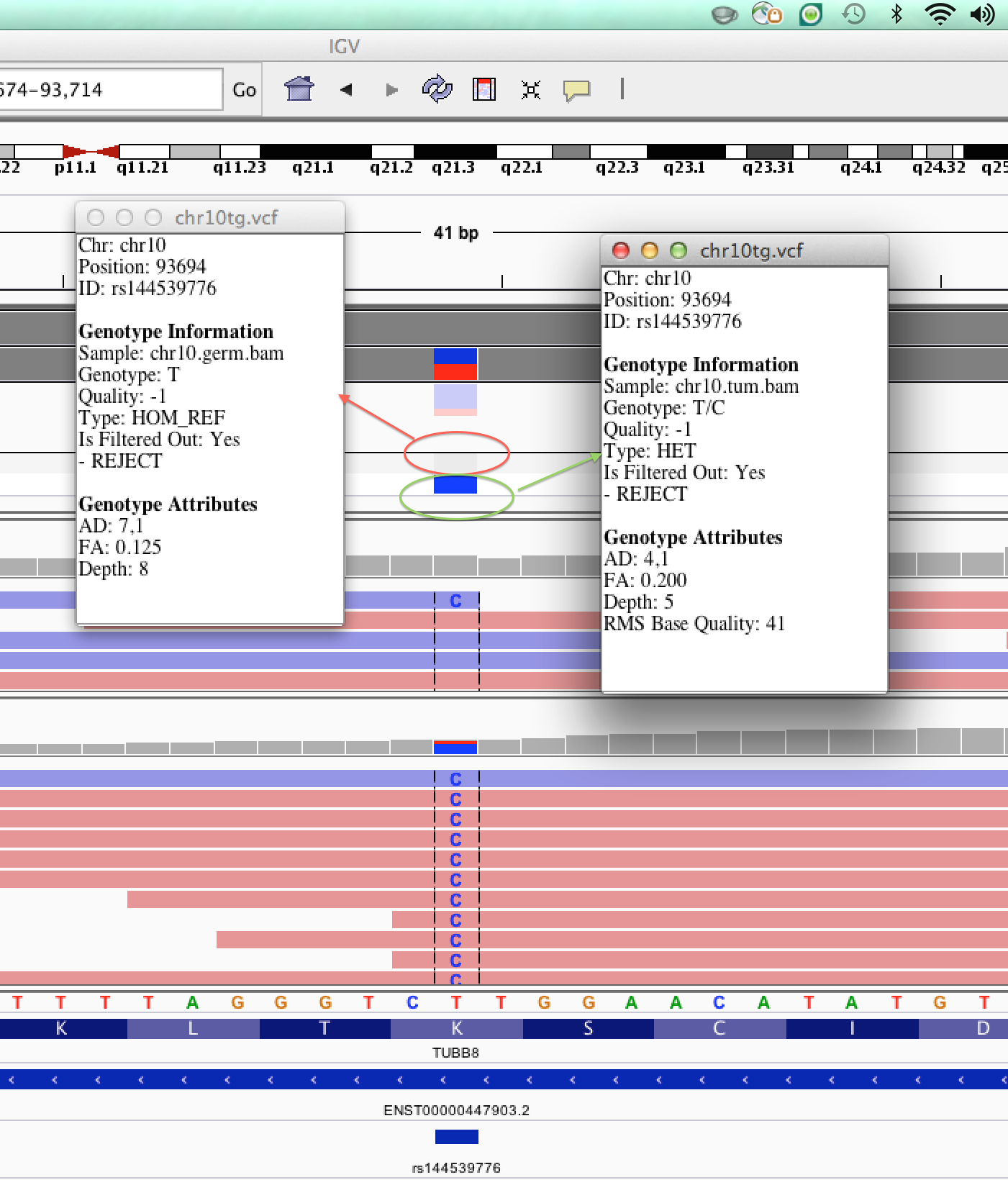
| chr10tg.vcf | Tumor-normal comparison vcf produced by somatic variant caller (MuTect) | vcf |
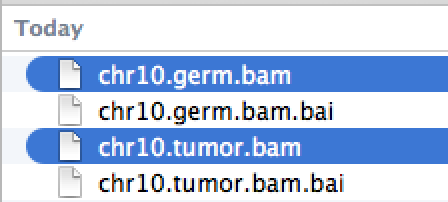
| chr10.germ.bam | Alignment file from germline (normal) sample | bam |
| chr10.tumor.bam | Alignment file from tumor sample | bam |
To load vcf and bam files, you need index files [".idx" (for vcf) or ".bai" (for bam)] to be in the same
directory
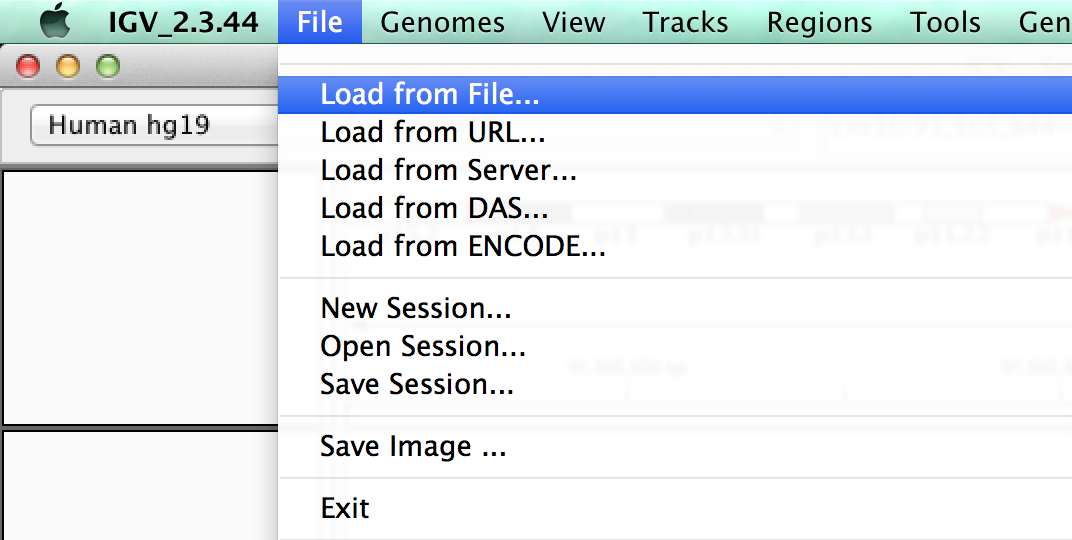
2. You can use a URL: click on load from URL - the files are in a publicly accessible folder in a remote server (helix)
Copy and paste the following URL's one at a time into the box (ctrl-c, ctrl-v):
http://helix.nih.gov/~CCBR/exome/chr10g.vcf
http://helix.nih.gov/~CCBR/exome/chr10t.vcf
http://helix.nih.gov/~CCBR/exome/chr10tg.vcf
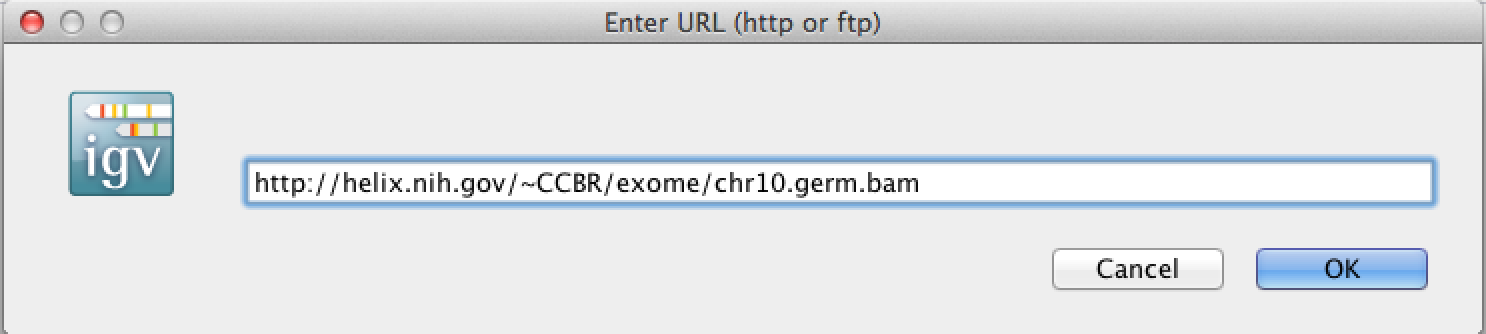
http://helix.nih.gov/~CCBR/exome/chr10.germ.bam
http://helix.nih.gov/~CCBR/exome/chr10.tumor.bam
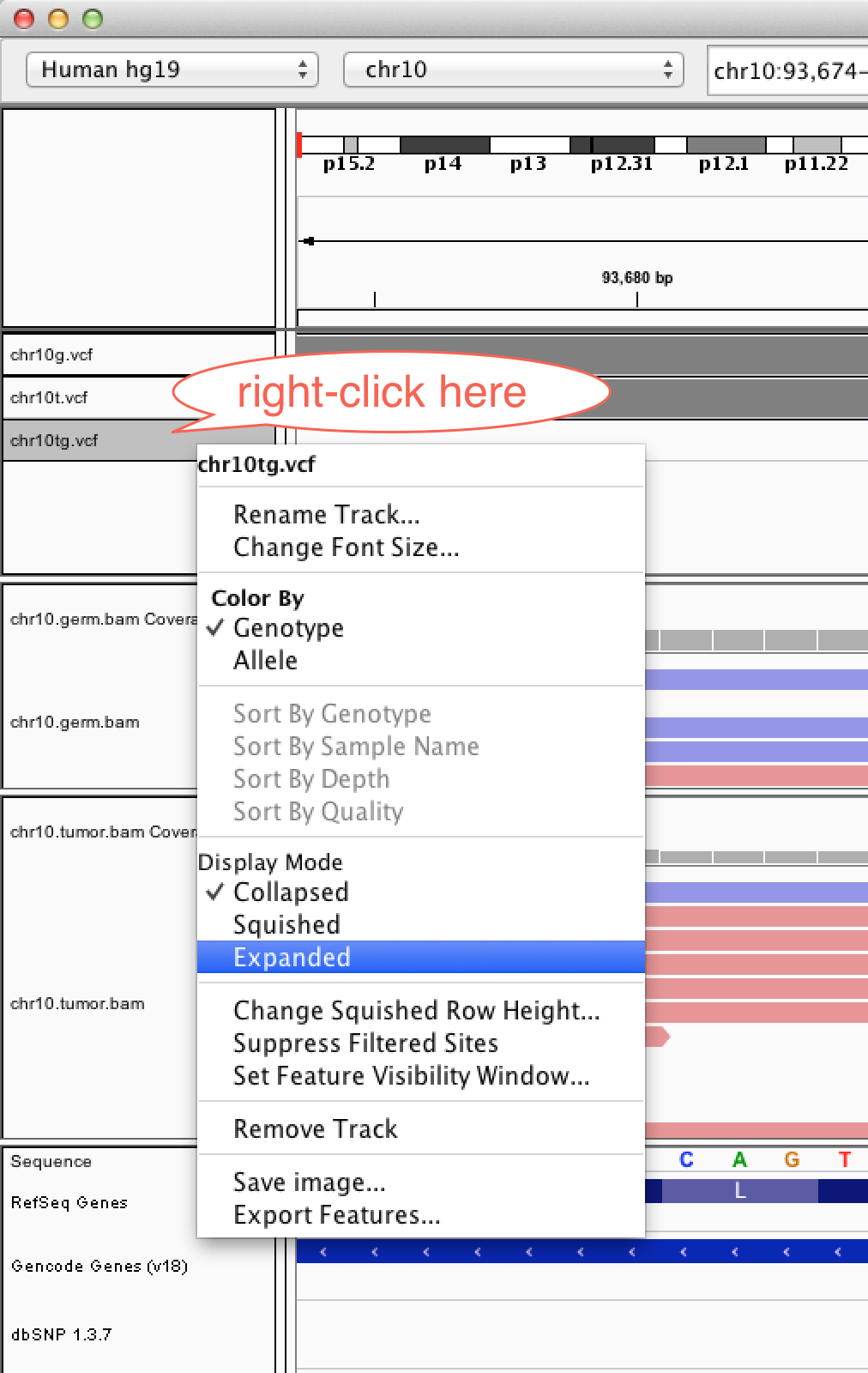
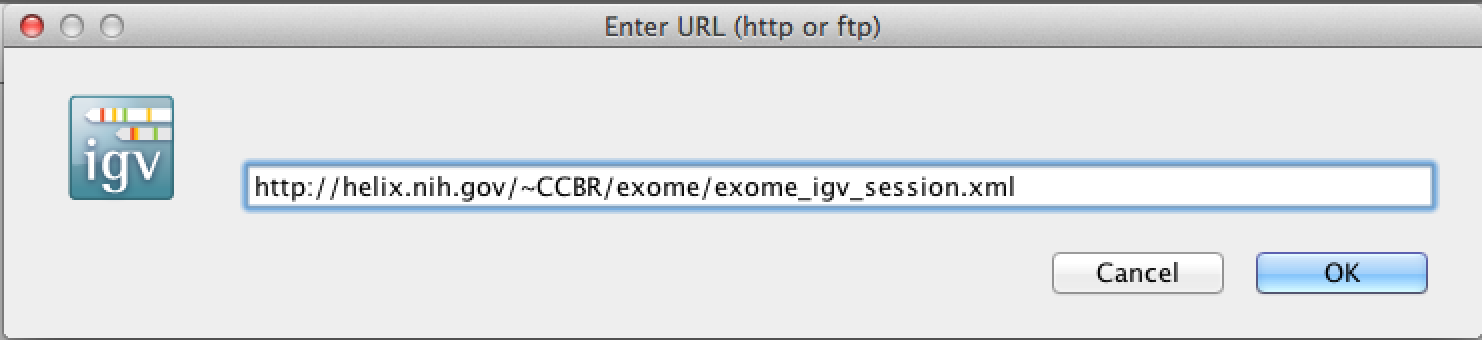
3. You can load an entire saved session by opening a local xml file (using Open Session), or in this case, using a URL as before
Copy and paste the following into the URL box :
http://helix.nih.gov/~CCBR/exome/exome_igv_session.xml
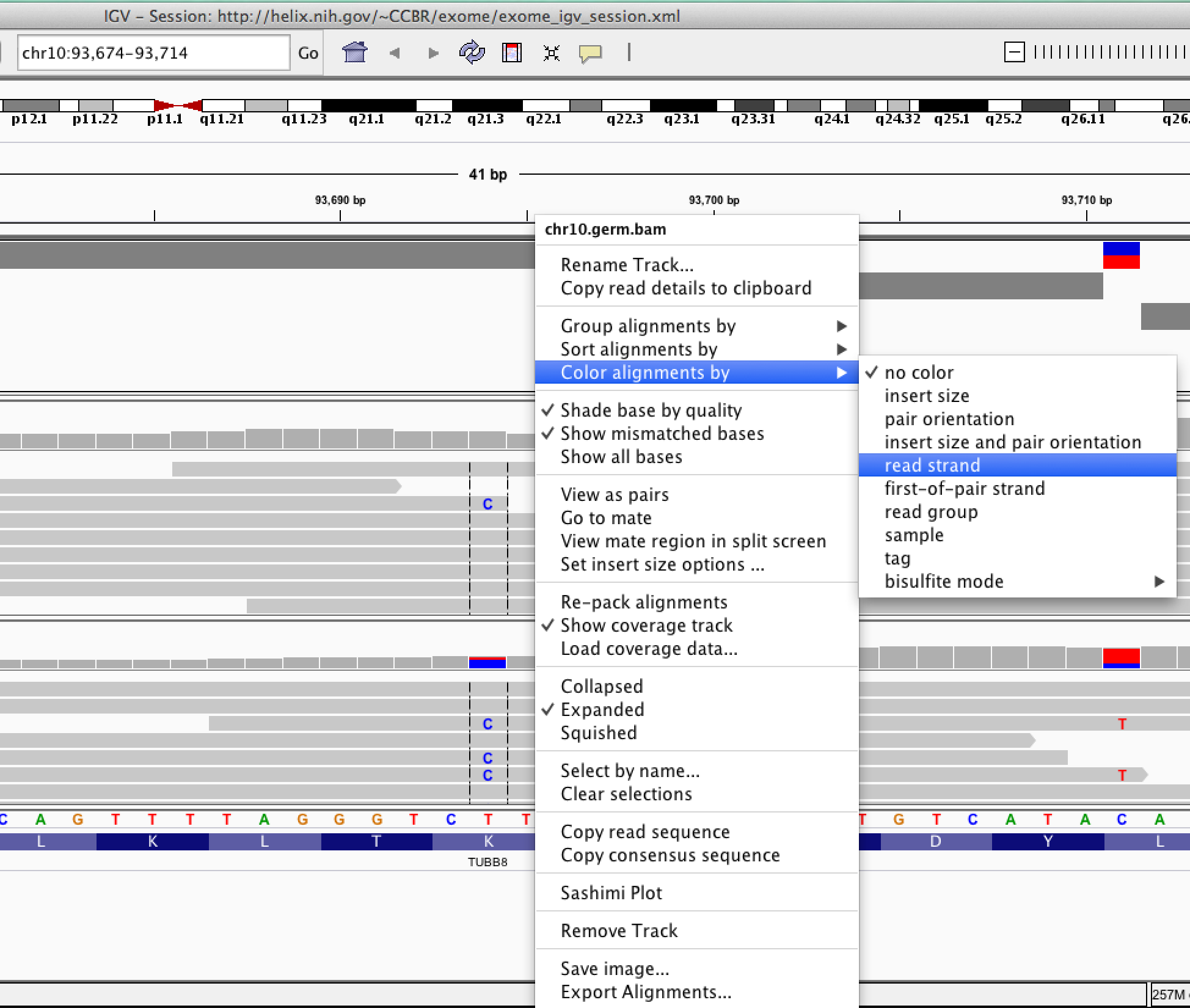
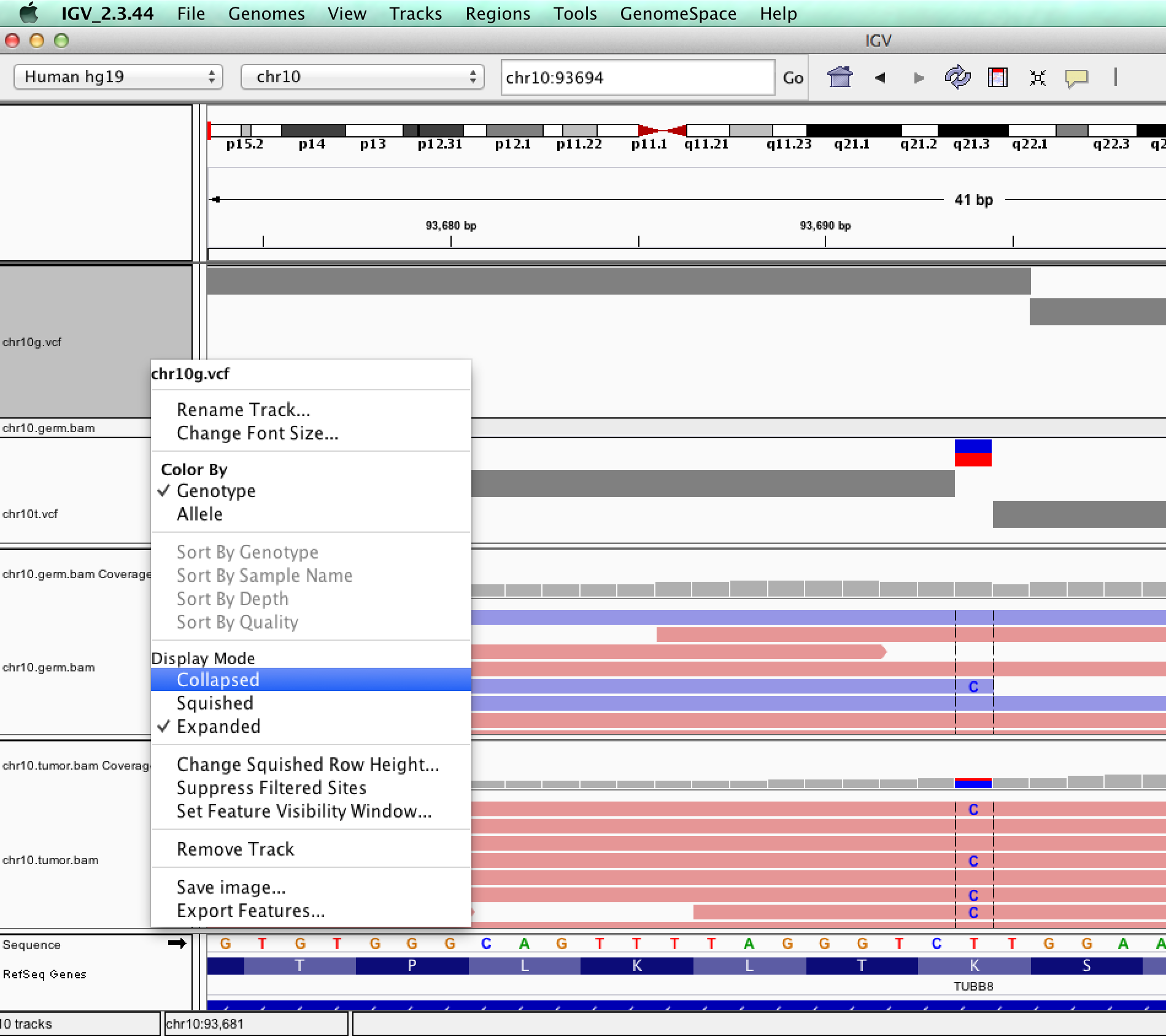
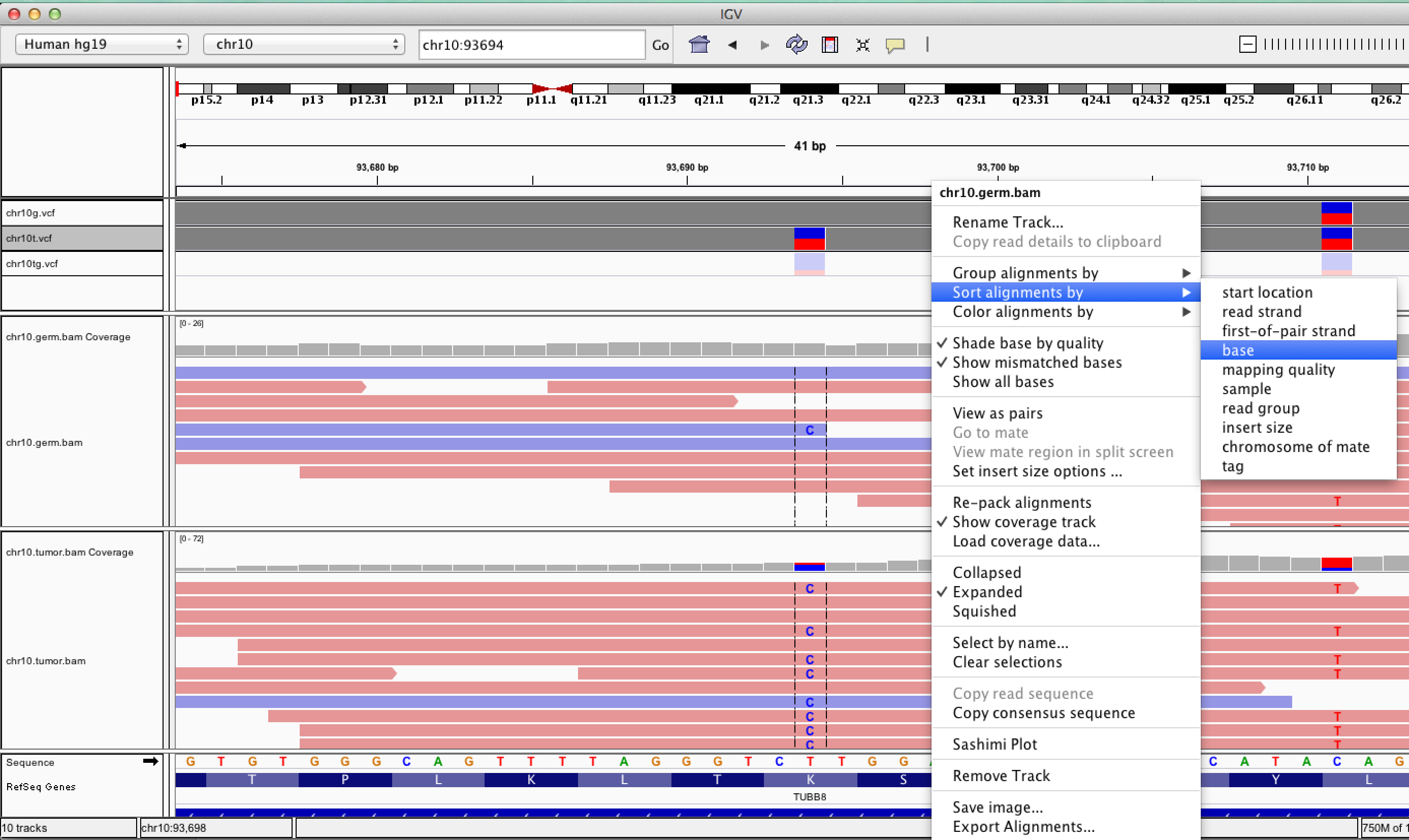
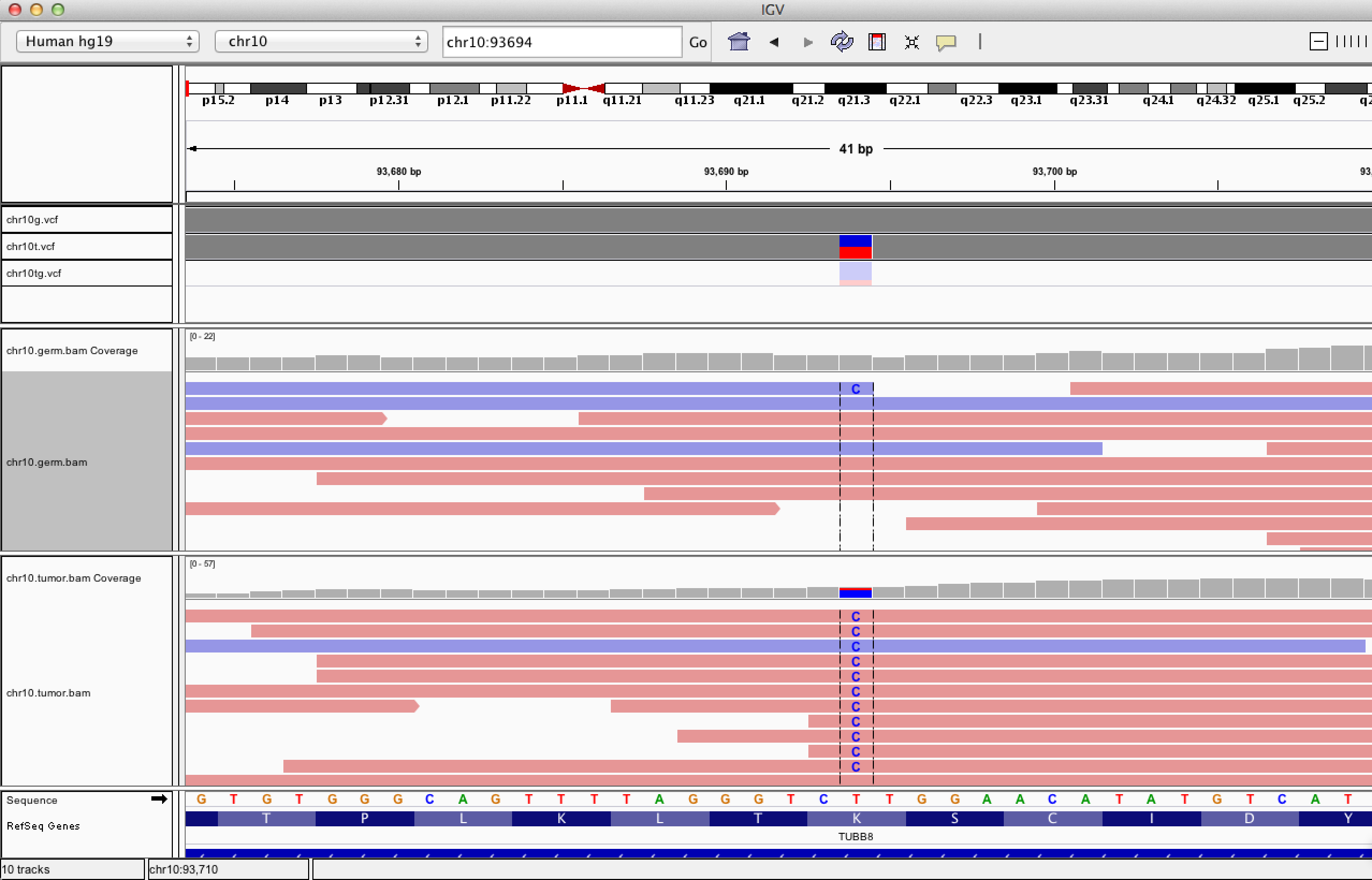
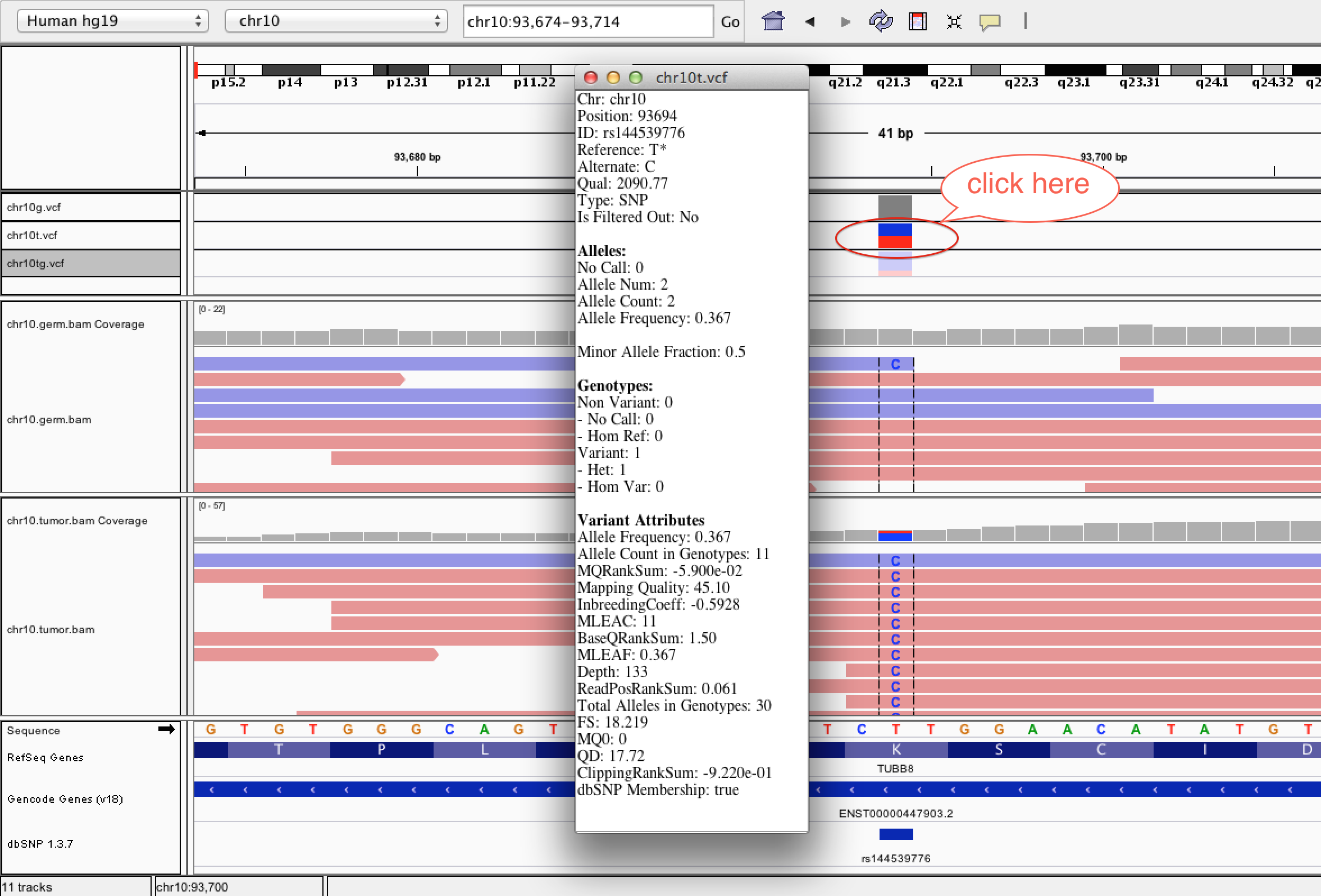
| ID | Description | Value |
|---|---|---|
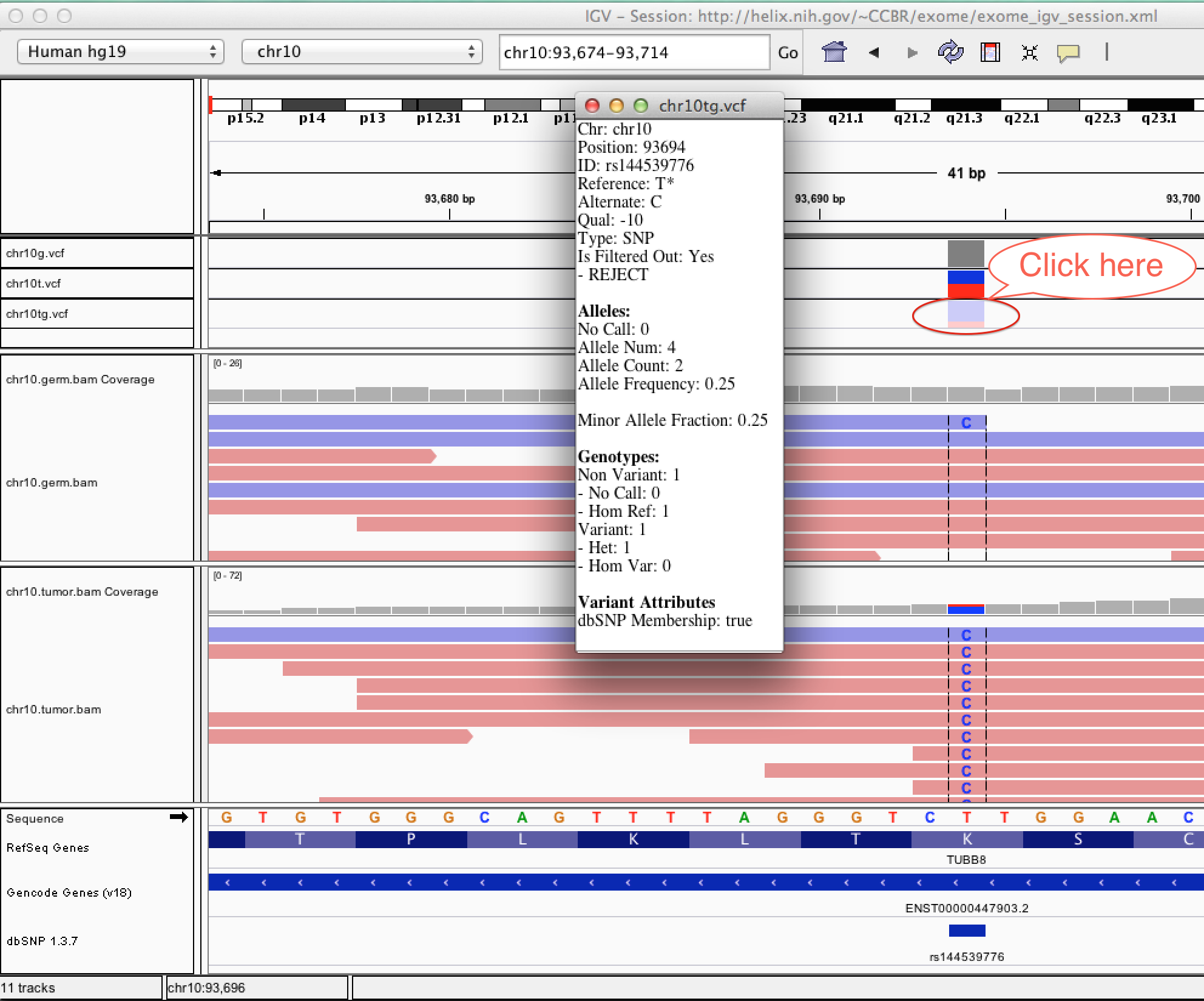
| AF | Allele Frequency, for each ALT allele, in the same order as listed | 0.367 |
| AC | Allele count in genotypes, for each ALT allele, in the same order as listed | 11 |
| MQRankSum | Z-score From Wilcoxon rank sum test of Alt vs. Ref read mapping qualities | -5.900e-02 |
| MQ | RMS Mapping Quality | 45.10 |
| MLEAC | Maximum likelihood expectation (MLE) for the allele counts (not necessarily the same as the AC), for each ALT allele, in the same order as listed | 11 |
| BaseQRankSum | Z-score from Wilcoxon rank sum test of Alt Vs. Ref base qualities | 1.50 |
| MLEAF | Maximum likelihood expectation (MLE) for the allele frequency (not necessarily the same as the AF), for each ALT allele, in the same order as listed | 0.367 |
| DP | Approximate read depth; some reads may have been filtered | 133 |
| ReadPosRankSum | Z-score from Wilcoxon rank sum test of Alt vs. Ref read position bias | 0.061 |
| AN | Total number of alleles in called genotypes | 30 |
| FS | Phred-scaled p-value using Fisher's exact test to detect strand bias | 18.219 |
| MQ0 | Total Mapping Quality Zero Reads | 0 |
| QD | Variant Confidence/Quality by Depth | 17.72 |
| ClippingRankSum | Z-score From Wilcoxon rank sum test of Alt vs. Ref number of hard clipped bases | -9.220e-01 |
| DB | dbSNP Membership | Flag |